|
|
You are here: Foswiki>DILIS Web>ResearchProjects (25 Feb 2025, Phiort1UserTopic)Edit Attach
DILiS - Research Projects
DrDimont: Explainable drug response prediction from differential analysis of multi-omics networks
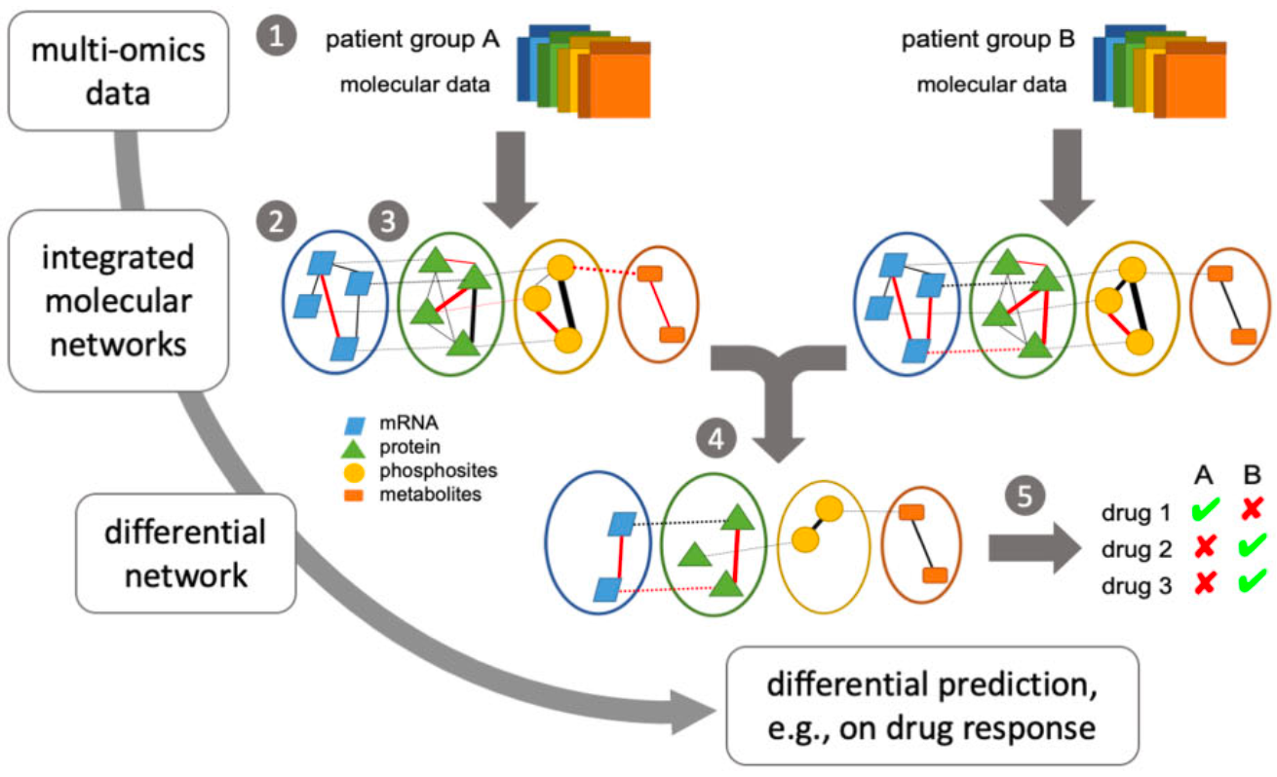 We developed a novel pipeline, DrDimont, that predicts the differential drug response of two different phenotypes, e.g., cancer subtypes, employing multi-omics networks. Our biological networks include data from gene expression analyses, protein quantifications, and metabolite quantifications. With DrDimont, two multi-layer networks are constructed for the two different conditions based on the correlation of the respective biological omics level. After combining the two networks to a differential network, the drug response is predicted. The differential drug response indicates whether a drug might have a different effect in the two groups. As a case study, we analyze the differential drug response in breast cancer data of ER+ (Estrogen Receptor positive) and ER- (Estrogen Receptor negative) patients. The tool DrDimont is available on CRAN and results have been published here.
We developed a novel pipeline, DrDimont, that predicts the differential drug response of two different phenotypes, e.g., cancer subtypes, employing multi-omics networks. Our biological networks include data from gene expression analyses, protein quantifications, and metabolite quantifications. With DrDimont, two multi-layer networks are constructed for the two different conditions based on the correlation of the respective biological omics level. After combining the two networks to a differential network, the drug response is predicted. The differential drug response indicates whether a drug might have a different effect in the two groups. As a case study, we analyze the differential drug response in breast cancer data of ER+ (Estrogen Receptor positive) and ER- (Estrogen Receptor negative) patients. The tool DrDimont is available on CRAN and results have been published here.
Variance feature attribution (VFA)
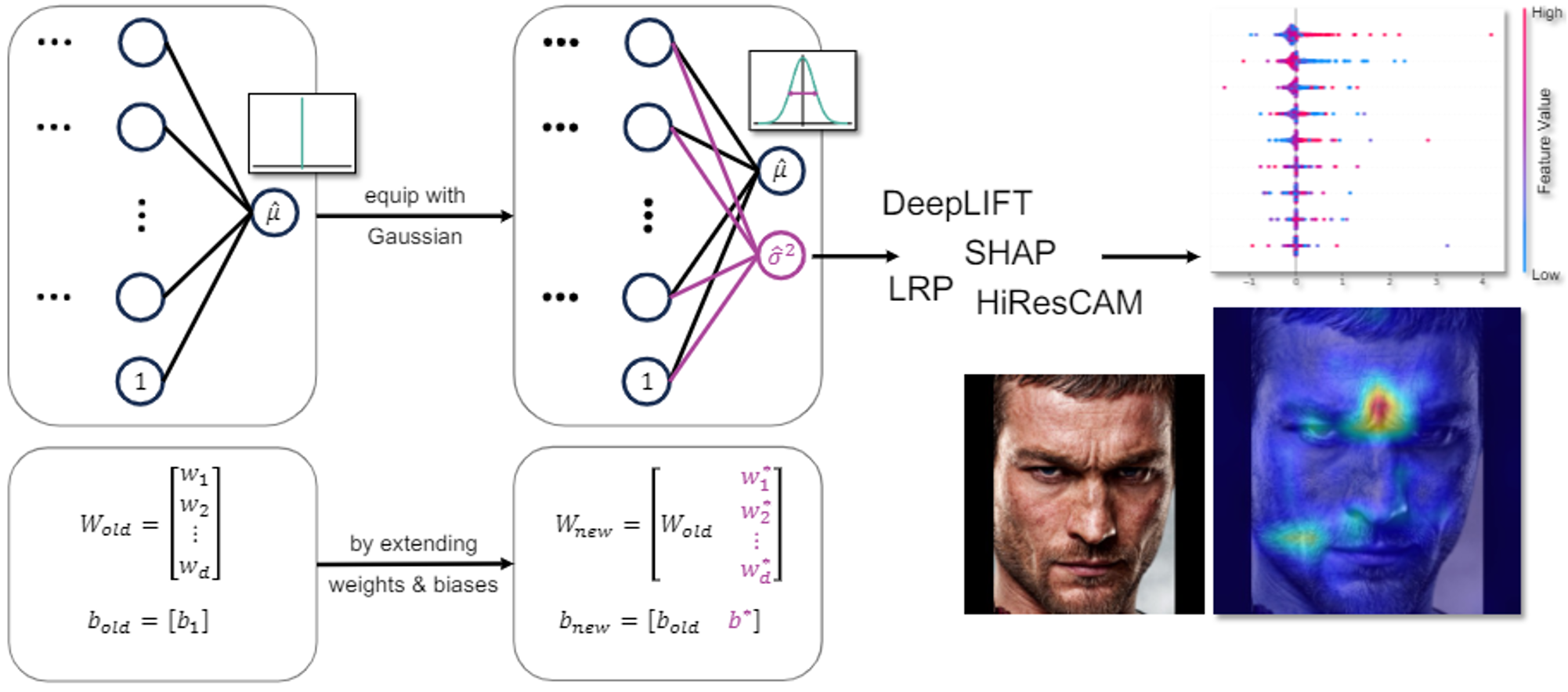 Explainability and uncertainty quantification are two pillars of trustable artificial intelligence. However, the reasoning behind uncertainty estimates is generally left unexplained. Understanding AI predictions should involve not just the output but also comprehending the underlying uncertainty, crucial for trust. We study methods of explaining the source behind uncertainty to enhance comprehension and trust in AI predictions. This knowledge can be utilized to formulate hypotheses to improve the model or to detect unintended shortcuts in the uncertainty estimation process, such as spurious correlations or biases. We are particularly interested in applying these methods to biomedical datasets. In healthcare, trustworthy AI, marked by transparent predictions and uncertainties, is essential for informed decision-making and fostering confidence among practitioners and patients. See Iversen and Witzke et al., 2023, for a preprint.
Explainability and uncertainty quantification are two pillars of trustable artificial intelligence. However, the reasoning behind uncertainty estimates is generally left unexplained. Understanding AI predictions should involve not just the output but also comprehending the underlying uncertainty, crucial for trust. We study methods of explaining the source behind uncertainty to enhance comprehension and trust in AI predictions. This knowledge can be utilized to formulate hypotheses to improve the model or to detect unintended shortcuts in the uncertainty estimation process, such as spurious correlations or biases. We are particularly interested in applying these methods to biomedical datasets. In healthcare, trustworthy AI, marked by transparent predictions and uncertainties, is essential for informed decision-making and fostering confidence among practitioners and patients. See Iversen and Witzke et al., 2023, for a preprint.
Drug response and uncertainty
Due to the complexity of biological systems, finding the right treatment for a specific disease can be challenging. Computational models can help our understanding of the effects of drugs and make them amenable to analysis. Thus, we build computational representations of these complex biological systems by modeling them as large networks that capture the interactions between the molecules in cells. We use network science and machine learning approaches to analyze these networks. For example, we employ graph neural networks to learn meaningful patterns that can help in predicting treatment outcomes. We aim to consider and combine the multiple layers of information that are obtained for different types of molecules, such as proteins and mRNAs. These layers include data-driven information like molecule measurements and prior knowledge information, for example, protein-interaction networks. Since many diseases are treated with multiple drugs at the same time, we are also working on building a framework to computationally analyze the response to multiple drugs. The presence of noise is an inherent characteristic of biological systems. Thus, we are leveraging uncertainty estimation methods to enhance the reliability and robustness of our predictions. This is not only essential for refining our computational models but also crucial in transitioning toward real-world applications where reliable uncertainty estimates are paramount for effective decision-making in disease treatment strategies.
In the future, we plan to bridge the gap between molecular predictions and clinically relevant predictions for patients by leveraging data from electronic health records (collaboration with the Hasso Plattner Institute at Mount Sinai, Icahn School for Medicine at Mount Sinai, USA).
In the future, we plan to bridge the gap between molecular predictions and clinically relevant predictions for patients by leveraging data from electronic health records (collaboration with the Hasso Plattner Institute at Mount Sinai, Icahn School for Medicine at Mount Sinai, USA).
Informing machine learning with simulations from dynamical models
Machine learning (ML) has become indispensable for data analysis, and especially of molecular data, such as (multi-)omics in the biomedical field. However, for molecular data, coherent datasets for specific contexts, conditions, and prediction tasks, especially including many samples, are still sparsely available. Difficulties resulting from learning on small datasets can be overcome by methods of informed machine learning, a recent branch of ML research in that available prior knowledge is used in combination with raw data. Throughout life sciences and especially within molecular biology, substantial research effort has been invested to curate in-depth information. There is knowledge on the complex structure and dynamics of central signaling pathways and cellular metabolism, as well as on mechanistic principles such as in infection dynamics. Many of these insights are captured in differential equation models, but not yet fully used in the context of ML. We propose to leverage the knowledge from these models with SimbaML (simulation-based machine learning), our framework for generating realistic synthetic data and applying them in ML pipelines.
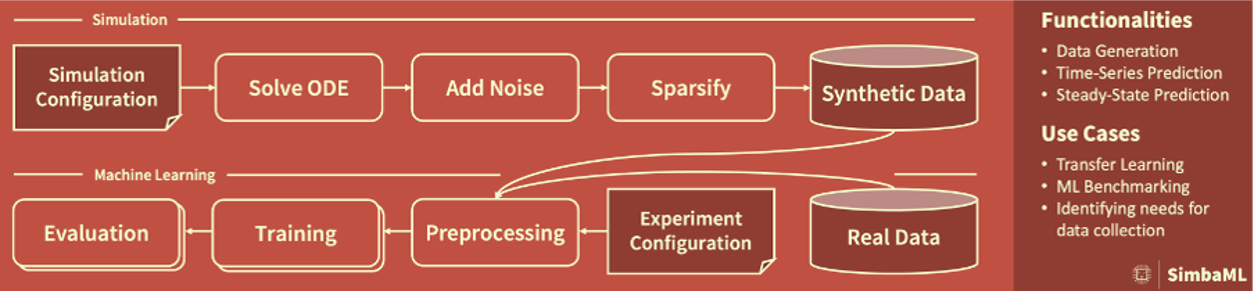
SimbaML is available as Python package (100% test coverage, compatible with scikit-learn, PyTorch, TensorFlow) from PyPI that you can use with straightforward config files to generate synthetic data and train and pretrain ML models, and check out our publication at ICLR here.
In addition, we investigated how synthetic datasets from ODE models should look like in order to make transfer learning on them successful. We investigate three example systems for time series prediction and propose an optimization pipeline. The preprint of this pipeline and results of the investigations is posted here.
In addition, we investigated how synthetic datasets from ODE models should look like in order to make transfer learning on them successful. We investigate three example systems for time series prediction and propose an optimization pipeline. The preprint of this pipeline and results of the investigations is posted here.
Act-i-ML: Making informed machine learning active
In this BMBF-funded project that we tackle jointly with the Hasso-Plattner-Institute for Digital Engineering, we aim to develop methods for robust and resilient AI. The focus of the project is to make informed machine learning active and thus inlcude how to leverage small data best for learning. We aim to apply this to multiple fields, ranging from epidemic forecasting to semiconductor ion fluxes, and potentially traffic. Find more details on the project here.
Edit | Attach | Print version | History: r7 < r6 < r5 < r4 | Backlinks | View wiki text | Edit wiki text | More topic actions
Topic revision: r7 - 25 Feb 2025, Phiort1UserTopic
- This page was cached on 12 Mar 2025 - 04:19.
Ideas, requests, problems regarding Foswiki? Send feedback